Endothelial inflammation and dysfunction in COVID-19
Abstract
Summary: The biggest challenge in the COVID-19 pandemic besides the spread of the SARS-CoV-2 virus is to reduce mortality rates. As the number of cases continues to rise and new variants, some with at least partial resistance to vaccines, emerge, the need for better understanding of the underlying pathology of the disease and for improved therapeutic strategies grows urgently. The endothelium is a main target of most viral infections in the body. The dysregulation of the normal functions of endothelial cells (ECs) contributes greatly to the thrombo-inflammatory storm and subsequent blood clot associated deaths in COVID-19 patients. Therefore, in this review we emphasize on the importance of ECs in healthy resting state and in inflammation. We summarize the current understanding of SARS-CoV-2 pathogenicity and the key contributions of in vitro cell culture models some of which have established the ACE2 (angiotensin-converting enzyme 2) receptors as the main gates for viral entry in the cell. Lastly, we focus on 3D biofabrication methods for the design of better in vitro models that mimic the host environment including interactions of multiple cell types, simulation of blood flow and real-time viral infections. The development and implementation of such experimental platforms are critical to elucidate host-pathogen interactions and to test new antiviral drugs and vaccines in a controlled, safe, and highly reproducible and predictive manner.
Introduction
Inflammation is the result of the body’s innate immune response to invading agents such as foreign objects, chemicals or pathogens. Endothelial cells (ECs) are active participants and regulators of inflammation. When microorganisms like viruses invade host cells, different effectors of the innate immune system detect and attack the infected tissue [1]. Macrophages recognize invading pathogens or cell damage and are activated directly by pathogen-associated molecular patterns (PAMPs) or indirectly by damage-associated molecular patterns (DAMPs) [2]. As a result, a signaling cascade is initiated leading to the release of factors which recruit more leukocytes to the site of infection [3]. When macrophages recognize and engulf pathogens, TNF-α or IL-1 are released which induces ECs to present E-selectin, ICAM-1 and VCAM-1 on their surface. These adhesion molecules can interact with circulating leukocytes in the blood. Moreover, once activated, ECs synthesize and display chemokines on their luminal surface [4]. Consequently, circulating leukocytes contact ECs and attach rapidly, but with low affinity, to the upregulated E-selectins or VCAMs and roll on the endothelial surface [5]. The interaction between the corresponding receptors on immune cells and the cytokines released by ECs triggers an inflammatory signaling cascade on a positive feedback loop [6]. Lastly, leukocytes extravasate through the tight junctions between ECs and enter the pathogen-invaded tissue causing inflammation which further activates the endothelium [4]. Smooth muscle relaxation and vasodilation promote blood flow, increasing vascular leakage of plasma proteins (and likely viruses too) through the junctions into the infected tissues. Long-lasting endothelial inflammation, as seen in autoimmune conditions, can affect ECs and lead to dysregulated blood clotting [7].
When the endothelium is at its normal resting and functional state, it controls blood flow, vessel permeability, and suppresses inflammation. Importantly, ECs can also prevent coagulation by inhibiting the initiation phases of this process by binding and displaying tissue factor pathway inhibitors (TFPIs) and also by blocking of the factor VIIa-tissue factor complex [8].
Even if COVID-19 is considered mainly a disease of the respiratory system, there is piling evidence suggesting that the primary and perhaps most significant target of the SARS-CoV-2 virus is the endothelium [9]. Therefore, inflammation and EC activation are the immediate outcomes during infection with this pathogen. The consequences of endothelial dysfunction in COVID-19 range from disruption of the blood-air barrier in the lungs and acute respiratory disease syndrome (ARDS) to micro blood clots, diffuse coagulopathy and multiple organ failure [10, 11]. The aim of this review is to summarize our current understanding of the role of endothelial inflammation in COVID-19 pathology and to give a perspective on the importance of in vitro experimental platforms in the fight against the pandemic.
Endothelial inflammation in COVID-19 and associated complications
ECs and lung complications in COVID-19
The most severe fatal cases with COVID-19 are either due to an acute lung injury (ALI), and more specifically acute respiratory distress syndrome (ARDS), or to myocarditis combined with ARDS, both following bilateral interstitial pneumonia. It has recently become evident that these conditions are the result of severe damage not only of lung alveolar epithelia, but also of the lung capillary endothelia at the air-blood barrier. Other types of blood vessels like the large veins and renal vessels can also be damaged by SARS-CoV-2. Thereby, vasculitis and vascular thrombosis are encountered as a common post mortem pathological finding in severe COVID-19 patients [12].
Infection with SARS-CoV-2 is known to induce a pro-inflammatory cytokines storm, specifically elevating the concentrations of IL-1b, IL-6, and TNF. This phenomenon has been shown to play an important role in the progression of the tissue inflammation causing ARDS, and often leading to death [13]. ALI and ARDS are manifested by pathological events such as the recruitment of inflammatory and phagocytic cells, activation of the complement system and opsonization. Importantly, the subsequent increased permeability of the endothelium causes disruption of the air-blood barrier and accumulation of protein-rich fluid in the alveoli, as well as other systemic and hemodynamic effects [13]. Accordingly, the liquid that accumulates in the lungs due to leaky blood vessels displaces the air inside the alveoli and increases the area density observed by computer tomography (CT) scans. This pathological event known as pulmonary ground-glass opacity (GGO) has become perhaps the most characteristic feature of COVID-19 pneumonias. Importantly, the link between GGO and disruption of the air-blood barrier induced by SARS-CoV-2 has been described by several studies [14, 15]. Furthermore, markers of inflammation (IL-2) and endothelial damage (sFLT-1) have been shown to correlate strongly to GGO [16, 17]. Consistent with such cytokine-mediated immunopathology, elevated IL-1β, IL-6, and TNF are also observed in the broncho-alveolar lavage and plasma of ARDS patients [13]. Moreover, a positive correlation between serum level of these cytokines and mortality rate in ARDS patients is shown [13].
SARS-CoV-2 entry in ECs
It is well recognized that the normal regulatory function of ECs and their supporting pericytes contributes significantly to the maintenance of a controlled inflammatory response [18]. ECs secrete soluble cellular mediators like Von-Willebrand factor (VWF) factor, E- and P-selectin, intercellular adhesion molecules (ICAMs) and vascular adhesion molecules (VCAMs) [5]. They also produce proinflammatory mediators and chemoattractants. However, when the normal endothelial function is compromised, its regulatory role is impaired too. This is the case when SARS-CoV-2 enters the cells through binding of its spike (S) protein to the angiotensin-converting enzyme (ACE2) receptors, which are highly expressed on ECs (Figures 1A and 1B). The attachment of the S1 subunit of the S-protein to this receptor facilitates the engulfment of the viral particle. After ACE2 is engaged for initial entry into ECs, the virus also hijacks the transmembrane protease serine 2 (TMPRSS2) which is employed in priming and cleaving of the S-protein at the S1/S2 cleavage site. This in turn allows for the fusion of cellular and viral membranes so the virus is released into the cytosol, where it can replicate [19] (Figure 1C).
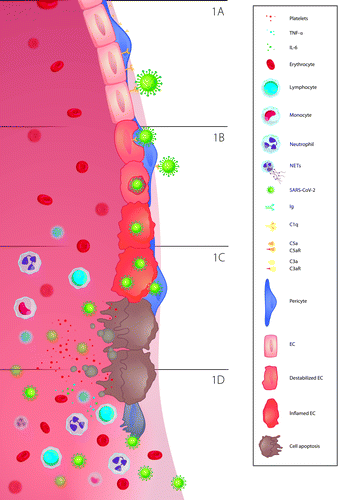
ECs are the primary target of SARS-CoV-2. The endothelium lines the lumen of the vasculature of the heart, lungs, kidneys, liver, and gastrointestinal tract making all these organs susceptible to the virus. Subsequently, apoptosis is triggered which leads to an inflammatory response cascade, ultimately recruiting more macrophages to the site of viral infection (Figures 1C and 1D). Different chemoattractants and cellular components are released from the dying infected cells, which further augments the adhesion of platelets and immune cells to the endothelial wall. While the inflammatory response is prominent, SARS-CoV-2 continues to invade other cells via the same mechanism and spreads into the tissues through weak EC tight junctions or through infected pericytes [20].
Effect of SARS-CoV-2 and complement activation on ECs
Several mechanisms of cell damage while fighting SARS-CoV-2 have been described. Once in the blood stream, the S-protein can be recognized and attached to circulating IgG antibodies (Figure 2A). Thereby, an antigen-antibody complex is formed, which can then be bound by the C1q proteins of the complement system (Figure 2B). The latter leads to activation of the classical complement pathway and the release of potent pro-inflammatory polypeptides like C5a and C3a, which recruit neutrophils as well as monocytes [11] (Figure 2B and 2C). Activation of neutrophils, in turn, brings forth the generation of extracellular traps (NETs). NETs contain C3, Properdin (P) and factor B, which activate the alternative complement pathway and set off an inflammatory feedback loop [21]. This in itself is enough to cause cell damage. C5a attracts leukocytes to the adjacent area. Furthermore, when C5a binds to its receptor, it primes leukocytes to attack the cell surface of the endothelium (Figure 2D). This also promotes upregulation and later degradation of vascular endothelial cadherin, which additionally destabilizes the cell and makes it ideal for leukocyte targeting [11].
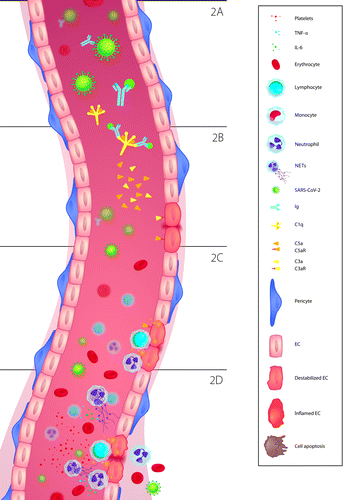
ECs and immunothrombosis
It has been well established that EC activation plays a role in the COVID-19 thrombo-inflammatory storm [22]. A high risk of venous as well as of arterial thrombosis with an overall 20% rate out of 8271 enrolled patients has been reported [23]. Potentially fatal complications like strokes and myocardial infarctions are also recognized due to dysfunctional endothelium [24]. ARDS is usually a manifestation of sepsis-induced organ dysfunction, characterized by endothelial barrier integrity disruption, an increased vascular permeability and leakiness, and diffuse lung damage. The imbalance between coagulation and inflammation is its predominant characteristic, resulting in an extreme inflammatory response, accompanied by diffuse fibrin deposition in the vascular capillary bed and alveoli [25].
Immunothrombosis, a key event in ARDS pathophysiology, is characterized by the recruitment and interaction of platelets and neutrophils at the site of endothelial injury. It is regulated by coagulation and inflammatory mediators, and is therefore considered as a humoral regulatory process, defined as “immunothrombosis” or “thromboinflammation” [25, 26]. Immunothrombosis results in the formation of an intravascular scaffold, enhancing the recognition and destruction of pathogens and supporting the endothelial integrity. The term “thromboinflammation” is initially used by Blair et al. in 2009 to describe their discovery that the activation of platelet toll-like receptor 2 (TLR2), a receptor best known for its role in triggering inflammation, also promotes thrombosis [27]. In 2013, Engelmann used the word “Immunothrombosis” to describe thrombosis “triggered by” or “involved with” innate immune responses [28]. Immunothrombosis in ARDS is accompanied by activated platelets, neutrophils, ECs, NETs, microparticles, and coagulation proteases. There is ample evidence that sepsis and ARDS are characterized by a procoagulant state, leading to a massive production of thrombin. The endothelial barrier is directly affected by thrombin, the predominant coagulation protein, which converts fibrinogen to fibrin. The diffuse alveolar and interstitial fibrin deposition induces the formation of microthrombi. Moreover, thrombin is the main activator of platelets, which further accumulate to the site of endothelial lesion, interacting with innate immune cells. In addition, the triggering of protease-activated receptors on immune cells by coagulation proteases induces pro- and anti-inflammatory reactions [25, 26, 29].
Thromboinflammatory storm in COVID-19
Although there is clinical evidence for SARS-CoV-2-induced thrombosis, the underlying mechanisms on the molecular level have not been entirely elucidated. Consistent evidence from COVID-19 patients shows that presence of the proinflammatory mediators TNF-α, IL-6 and fibrinogen strongly promotes the pro-coagulant state, which can lead to activation of thrombosis [30]. Both TNF-α and IL-6 were shown to possess a prothrombic effect in other infections such as sepsis and conditions like rheumatoid arthritis [31] or asthma [32]. These specific proinflammatory cytokines are elevated to a greater degree in SARS-CoV-2 infection compared to other diseases [30]. Interestingly, tissue-factor dependent thrombosis can be linked to SARS-CoV-2 infection. This is supported by emerging evidence that secretion of platelet tissue-factor VIIa from monocytes and macrophages leads to increased TNF-α activity [33]. What is possibly of even greater importance is the presence of circulating antiphospholipid antibodies, mainly anticardiolipin IgM, IgG and anti-phosphatidylserine/prothrombin (anti-PS/PT), which can activate ECs. The link between such autoantibodies and endothelial dysfunction is supported by several findings. Shi et al. have shown that serum from COVID-19 patients activates cultured ECs to express surface adhesion molecules with a well-established role in thrombosis such as E-selectin, ICAM-1 and VCAM. The authors have further demonstrated that there is a positive correlation between the presence of antiphospholipid antibodies and the upregulation of these endothelial activation markers. Furthermore, the addition of purified IgG antibodies from COVID-19 patient sera to control serum samples with depleted IgG could also induce ECs in vitro. Of note, endothelial activation could be mitigated by depletion of total IgG from the samples [24].
In some cases, a connection between circulation of antiphospholipid antibodies and the austerity of the SARS-CoV-2-driven thrombosis has been suggested. Such autoantibodies attach to cell surfaces and thus activate ECs, neutrophils, and platelets. It was found that COVID-19 IgG serum fractions, which are enriched for antiphospholipid antibodies, potently activate neutrophils in vitro and increase the chance for thrombosis when injected into mice [34]. All of these findings suggest an underlying mechanism which can explain the thrombo-inflammatory effects of SARS-CoV-2 [24].
Models and strategies for investigating endothelial inflammation and SARS-CoV-2 infection
In vitro models
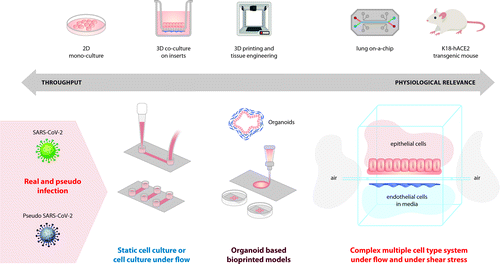
The most common models for studying disease pathogenicity and for pre-clinical evaluation of novel drugs and vaccines are associated with standard in vitro cell cultures and with animal in vivo research. While these two experimental platforms have their limitations and disadvantages, both modelling systems have already contributed greatly towards the understanding of COVID-19 pathogenesis. Even though not representative of the complex cell interactions in vivo, most current approaches for studying diseases, including viral infections, are based on conventional monolayer two-dimensional (2D) cell cultures. Compared to in vivo models, 2D cultures are inexpensive, easier to analyse and could be used for high-throughput SARS-CoV-2 drug screenings but may also lack predictivity. For instance, work in over a dozen commonly used human cell lines demonstrated that the spike (S) protein of SARS-CoV-2 shares sequence similarity to that of SARS-CoV and uses the same way of cell entry – through the ACE2 receptor. The significance of the serine protease TMPRSS2 for cell infection was also confirmed through in vitro studies. Importantly, Hoffman et al. showed that viral entry into cell lines can be effectively blocked by either antiserum against the ACE2 receptor and by the clinically approved protease inhibitor (camostat msylate) [19]. Similarly, human recombinant soluble ACE2 is shown to inhibit viral burden in an in vitro model of capillary organoids established from induced pluripotent stem cells [35]. However, such in vitro systems fail to mimic viral infection patterns, systemic responses and the complex cell-cell interactions that occur in the human body.
In vivo models
Over the past century the use of genetically established inbred mouse strains, knockout and more recently humanized mice have enabled the research community to advance the development of vaccines and antivirals against emerging infections [36]. The ongoing challenges of studying the pathogenesis of diseases and the high drug failure rates, however, reveal the limitations of in vivo models. A good illustration of the inherent disadvantages of animal models can be given by a SARS-CoV-2 infected transgenic mouse expressing the human ACE2 receptor under the cytokeratin-18 promoter (K18-hACE2). While good for studying lung immune infiltration and pathogenesis, the authors did not observe any signs of thrombosis, which is arguably the biggest threat to humans suffering from COVID-19 [37].
Thus, in vivo animal models lack high throughput capabilities and are not always representative of human biology. On the other hand, 2D cell cultures cannot accurately mirror the physiological effects of infections and the corresponding cell microenvironment. Therefore, new strategies to investigate SARS-CoV-2 pathogenicity and to establish alternative therapeutics are urgently needed [38, 39]. Three-dimensional (3D) biomodelling can bridge the gap between conventional cell cultures and in vivo models. Biofabrication techniques such as 3D bioprinting can produce living copies of tissues by high resolution layer-by-layer printing of cells and biochemicals embedded in biological materials (bioinks). Such technologies make it possible to create organoids with incorporated vasculature and microfluidic organ-on-chip platforms for in vitro recreation of the complex cell microenvironment [40].
Organ-on-Chip models
According to the definition elaborated by the Working group of EU initiative ORCHID (Organ-on-Chip development), “An Organ-on-Chip (OoC) is a fit for purpose fabricated microfluidic-based device, containing living engineered organ substructures in a controlled micro- or nanoenvironment that recapitulates one or more aspects of the dynamics, functionality and (patho)physiological response of an organ in vivo, in real-time monitoring mode” [41]. Mimicking organ environment, OoC has the ability to regulate key physiological parameters, including concentration gradients, shear force, cell patterning, tissue-boundaries, and tissue–organ interactions [42]. Many OoC recreating either micro-capillary networks or mimicking lung alveolar–capillary barrier are created. A recent OoC modelling complex vascular networks is based on a poly-dimethysiloxane (PDMS) microfluidic chip, where a 3 μm porous barrier separates endothelial from epithelial channels. The network is created in such a way that it provides high flow and low flow areas, generating heterogenous solute dissociation from ECs to epithelial compartments. It helps the establishment of an in vivo-like vascular morphology with fully enclosed lumen, thus generating heterogeneous epithelial phenotypes and modelling physiological microvascular environment shear stress [43]. Such OoC can be used to study cell-cell interactions and to assess the vascular inflammation rate by real-time measuring of the rolling, adhesion, and migration of immune cells towards ECs. Adding another level of complexity, several advanced OoCs have been developed for lung liquid-air interface mimicking that resemble native alveolar physiology. The most intricate one implements the same PDMS-based soft lithography approach as described above. It possesses 2D PDMS air and liquid chambers, covered by alveolar epithelial cells and human pulmonary microvascular ECs respectively. They are separated by a 10 μm thick semi-permeable membrane covered with extracellular matrix allowing for cell trans-migration [44]. In this model the structures of the membranes are altered under a vacuum to simulate expansion/contraction of the alveoli during respiration. Using lung-on-a-chip approach, SARS-CoV-2 viral infection models are established for assessing strain-dependent virulence, cytokine production and the recruitment of circulating immune cells, allowing to study viral pathogenesis and to test various drug candidates in in vivo-like conditions. This approach showed at gene expression, cell morphology and cell functionality levels that the air-liquid interface is imperative to recreate lung physiological conditions [45, 46]. Other models also support air-liquid interface, but prevent cell trans-migration using 2 μm pore membranes. These models also provide ciliated cellular phenotype of alveolar epithelia as well as functional tight-junctions in both epithelial and endothelial layers [47, 48]. Most recent models extended the concept by generating 3D GelMA (methacrylated gelatin hydrogel) alveoli like structures covered with epithelial and endothelial cells, combined with microfluidics in a chip device to provide breathing stretching of the entire structure. This approach resulted in an even more physiological like model and gave additional evidence supporting the ability of SARC-CoV-2 to infect ECs via ACE2 receptors [49].
Intricate proof-of-concept microfluidics OoC alveolar models have been developed in the past and have shown promising ability to mimic the physiology of the lungs in health and disease (such as asthma) [50]. Of note, perhaps the most elaborate experimental 3D biofabricated system used to study SARS-CoV-2 has combined primary pulmonary epithelial and ECs in an airway-on-a-chip device consisting of two channels – one exposed to air where cells would be subjected to viruses mimicking air-borne infection, and a microfluidics channel where medium would be supplied together with therapeutic agents. Using this intricate in vitro model Si et al. showed not only excellent morphological and transcriptional replication of in vivo air-blood barriers, but also that such systems allow for rapid screens of anti-viral drugs which may be effective for COVID-19 treatment [46]. Implementing a similar lung-on-a-chip model, Thacker and colleagues managed to monitor and elucidate certain aspects of SARS-CoV-2 pathogenicity. They found persistent endothelial inflammatory response compared to a more transient epithelial one, air-blood barrier permeability changes (due to loss of tight junctions), endothelial damage. It is demonstrated in vitro that infection with this virus can induce a pro-coagulatory transcriptional program in blood vessels [51].
Biofabrication in vascular biology of COVID-19
Biofabrication techniques offer many advantages for studying virus-host interactions similar to those occurring in vivo, but in a tightly controlled, reproducible, and high throughput manner. Most importantly, physiologically-relevant conditions for the cells are produced via such methods, which allows for a more realistic assessment of new anti-viral drugs, different modes of infection, host-pathogen interactions, and even make it possible to culture otherwise non-cultivable viruses [52]. The relevance of 3D bioprinting in viral research is demonstrated recently by a study using an alveolar cell line (A549) infected with Influenza A virus. Berg et al. showed a physiological pattern of clustered infection of cells in their model and release of interferons – two key events not observed in standard monolayer cultures [53]. Even relatively simple 3D bioprinted methacrylated gelatin (GelMa) based matrices are shown to support the growth of standard cell lines to adapt tissue-like morphology of transitional epithelium and even of endothelium. Importantly, such an experimental platform has proven its capacity to be successfully infected and to allow monitoring of viral-host interactions [52].
Mirroring the complex air-blood barrier with alveolar cells and ECs is also made possible via 3D bioprinting. Horvath et al. demonstrated that the controlled deposition of these two cell types uniformly and in close proximity can replicate the morphology and certain functional aspects of the lung [54].
The more recent 3D pulmonary models are based on induced pluripotent stem cells and embryonic stem cells which allow viral replication in vitro and thereby the emulation of host immune responses [55]. Alveolar models, developed from ciliated and goblet cells, displayed a fast entry of SARS-CoV-2 and development of a subsequent infection in both cell types. Furthermore, infected HAT2 cells mounted a prominent pro-inflammatory response with upregulation of most Interferon signalling genes except Interferon β1 (IFN-β1) and Interferon 3 (IFN-3) [56]. The use of remdesivir was able to inhibit the viral replication at a greater rate when compared to IFN-β1 in alveolar cultures (AT2) [57]. This study managed to portray the importance of immune cells and their role in the alveolar barrier dysfunction caused by the expression of IL-6 during viral infection [58].
A summary of the available approaches for experimental modelling of SARS-CoV-2 infection is shown in Figure 3.
Conclusions
The importance of studying the endothelium is growing as SARS-CoV-2 continues to claim lives in spite of the ongoing global vaccination programmes. Furthermore, constantly emerging variants of the virus imply that research needs to continue and be ready to respond promptly with appropriate modelling systems to any new and potentially deadlier viruses. It is now established that SARS-CoV-2 infects ECs either through endocytosis or by infiltrating damaged ECs. In addition, the virus can be recognized by IgG antibodies and can form antigen-antibody complexes which activate the complement pathway and induce cell damage. As part of the pathology observed in COVID-19, ECs undergo a switch from an inactive phenotype to a state of self-defence, which however is dysfunctional. Thereby, the endothelium loses its ability to control the inflammatory and coagulation processes in the vasculature and allows the formation of potentially life-threatening blood clots.
Lack of quick and effective therapies makes fending off the infection difficult. With recent development of 3D bioprinting models for studying host-pathogen interactions and the inflammatory response, there are promising steps towards safe and reproducible in vitro systems for drug testing. Progress using microfluidics and organ-on-a-chip platforms has the potential to animate the interaction in the blood-endothelial network. This is possible thanks to the system’s ability to cultivate cells and blood from patients in near-physiological conditions. Bioengineering of microfluidic blood vessels can help elucidate the mechanism of blood clotting caused by SARS-CoV-2 [59]. In addition, fabrication of lung-on-a-chip models that mimic the environmental, structural, and functional intricacy in the native tissue are also developed. This offers an opportunity to thoroughly investigate host-pathogen interactions at the air-blood barrier. Recreating the mode of infection via in vitro biofabrication techniques will undoubtedly improve both the understanding of the pathogenesis of COVID-19 and the ongoing pre-clinical testing of more effective pharmacological compounds that will help fight the pandemic.
References
1 Inflammatory responses and inflammation-associated diseases in organs. Oncotarget. 2018;9(6):7204–18.
2 . Pattern recognition receptors and the host cell death molecular machinery. Front Immunol. 2018;9:2379.
3 . Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol. 2012;4(3):a006049.
4 . Linking endothelial dysfunction with endothelial cell activation. J Clin Invest. 2013;123(2):540–1.
5 .
Leukocyte-endothelial cell adhesion . In Granger DNSenchenkova E, editors. Inflammation and the microcirculation. San Rafael (CA): Morgan & Claypool Life Sciences; 2010.6 . The role of inflammatory cytokines in endothelial dysfunction. Basic Res Cardiol. 2008;103(5):398–406.
7 . Chronic inflammatory diseases and endothelial dysfunction. Aging Dis. 2016;7(1):81–9.
8 . Production and control of coagulation proteins for factor X activation in human endothelial cells and fibroblasts. Sci Rep. 2020;10(1):2005.
9 . Endothelial Injury in COVID-19 and Acute Infections: Putting the Pieces of the Puzzle Together. Arterioscler Thromb Vasc Biol. 2021;41(5):1774–6.
10 Vascular endothelial damage in the pathogenesis of organ injury in severe COVID-19. Arterioscler Thromb Vasc Biol. 2021;41(5):1760–73.
11 . Endothelial activation and dysfunction in COVID-19: from basic mechanisms to potential therapeutic approaches. Signal Transduct Target Ther. 2020;5(1):293.
12 Postmortem examination of COVID-19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings in lungs and other organs suggesting vascular dysfunction. Histopathology. 2020;77(2):198–209.
13 . Therapeutic potential of neurotrophic factors and neural stem cells against ischemic brain injury. J Cereb Blood Flow Metab. 2000;20(10):1393–408.
14 Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. 2020;383(2):120–8.
15 Pulmonary vascular manifestations of COVID-19 pneumonia. Radiol Cardiothorac Imaging. 2020;2(3):e200277.
16 . sFlt-1 and CA 15.3 are indicators of endothelial damage and pulmonary fibrosis in SARS-CoV-2 infection. Sci Rep. 2021;11(1):19979.
17 Correlation between ground-glass opacity on pulmonary CT and the levels of inflammatory cytokines in patients with moderate-to-severe COVID-19 pneumonia. Int J Med Sci. 2021;18(11):2394–400.
18 . Pericytes: Problems and Promises for CNS Repair. Front Cell Neurosci. 2019;13:546.
19 SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181(2):271–80.
20 . SARS-CoV-2 infects cells after viral entry via clathrin-mediated endocytosis. J Biol Chem. 2021;296:100306.
21 The complement system in COVID-19: friend and foe? JCI Insight. 2020;5(15):e140711.
22 Mechanisms of COVID-19 thrombosis in an inflammatory environment and new anticoagulant targets. Am J Transl Res. 2021;13(5):3925–41.
23 . Thromboembolism risk of COVID-19 is high and associated with a higher risk of mortality: A systematic review and meta-analysis. EClinicalMedicine. 2020;29:100639.
24 Endothelial cell-activating antibodies in COVID-19. medRxiv. 2021;21250041.
25 . Immunothrombosis in Acute Respiratory Distress Syndrome: Cross Talks between Inflammation and Coagulation. Respiration. 2017;93(3):212–25.
26 . The era of thromboinflammation: platelets are dynamic sensors and effector cells during infectious diseases. Front Immunol. 2019;10:2204.
27 Stimulation of Toll-like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3-kinase. Circ Res. 2009;104(3):346–54.
28 . Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. 2013;13(1):34–45.
29 . Blood coagulation in immunothrombosis-At the frontline of intravascular immunity. Semin Immunol. 2016;28(6):561–9.
30 . Thrombosis in COVID-19. Am J Hematol. 2020;95(12):1578–89.
31 Inflammatory and prothrombotic biomarkers in patients with rheumatoid arthritis: effects of tumor necrosis factor-alpha blockade. J Autoimmun. 2008;31(2):175–9.
32 Between inflammation and thrombosis – endothelial cells in COVID-19. Eur Respir J. 2021;58:2100377.
33 Activated monocytes enhance platelet-driven contraction of blood clots via tissue factor expression. Sci Rep. 2017;7(1):5149.
34 Prothrombotic antiphospholipid antibodies in COVID-19. medRxiv. 2020;20131607.
35 Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell. 2020;181(4):905–13.
36 . Mouse Models as Resources for Studying Infectious Diseases. Clin Ther. 2019;41(10):1912–22.
37 SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. Nat Immunol. 2020;21(11):1327–35.
38 Engineering a model to study viral infections: bioprinting, microfluidics, and organoids to defeat coronavirus disease 2019 (COVID-19). Int J Bioprint. 2020;6(4):302.
39 3D Bioprinting for fabrication of tissue models of COVID-19 infection. Essays Biochem. 2021;65(3):503–18.
40 3D tissue models as an effective tool for studying viruses and vaccine development. Front Mater. 2021;8(80).
41 Organ-on-chip in development: Towards a roadmap for organs-on-chip. ALTEX. 2019;36(4):650–68.
42 Organ-on-a-chip: recent breakthroughs and future prospects. Biomed Eng Online. 2020;19(1):9.
43 A Microvascularized Tumor-mimetic Platform for Assessing Anti-cancer Drug Efficacy. Sci Rep. 2018;8(1):3171.
44 . Reconstituting organ-level lung functions on a chip. Science. 2010;328(5986):1662–8.
45 Biomimetic human disease model of SARS-CoV-2 Induced lung injury and immune responses on organ chip system. Adv Sci. 2020;2002928.
46 A human-airway-on-a-chip for the rapid identification of candidate antiviral therapeutics and prophylactics. Nat Biomed Eng. 2021;5(8):815–29.
47 Small airway-on-a-chip enables analysis of human lung inflammation and drug responses in vitro. Nat Methods. 2016;13(2):151–7.
48 Co-cultured microfluidic model of the airway optimized for microscopy and micro-optical coherence tomography imaging. Biomed Opt Express. 2019;10(10):5414–30.
49 Reversed-engineered human alveolar lung-on-a-chip model. Proc Natl Acad Sci. 2021;118(19):e2016146118.
50 Development of a functional airway-on-a-chip by 3D cell printing. Biofabrication. 2018;11(1):015002.
51 . Rapid endotheliitis and vascular damage characterize SARS-CoV-2 infection in a human lung-on-chip model. EMBO Rep. 2021;22(6):e52744.
52 Simplified bioprinting-based 3D cell culture infection models for virus detection. Viruses. 2020;12(11):1298.
53 Optimization of cell-laden bioinks for 3D bioprinting and efficient infection with influenza A virus. Sci Rep. 2018;8(1):13877.
54 . Engineering an in vitro air-blood barrier by 3D bioprinting. Sci Rep. 2015;5:7974.
55 . Experimental models to study COVID-19 effect in stem cells. Cells. 2021;10(1):91.
56 . Human pluripotent stem cell-based organoids and cell platforms for modelling SARS-CoV-2 infection and drug discovery. Stem Cell Res. 2021;53:102207.
57 SARS-CoV-2 infection of primary human lung epithelium for COVID-19 modeling and drug discovery. Cell Rep. 2021;35(5):109055.
58 . Antiviral treatment of COVID-19: An update. Turk J Med Sci. 2021;51:3372–90.
59 . Bioengineered in vitro tissue models to study SARS-CoV-2 pathogenesis and therapeutic validation. ACS Biomater Sci Eng. 2020;6(12):6540–55.